2014 is the International Year of Crystallography; a celebration of X-ray crystallography and the science driven by this technology. 2014 also marks 10 years since Griffith University solved its first structure of a protein, a world-first discovery of the structure of a protein essential for the debilitating rotavirus to infect cells.

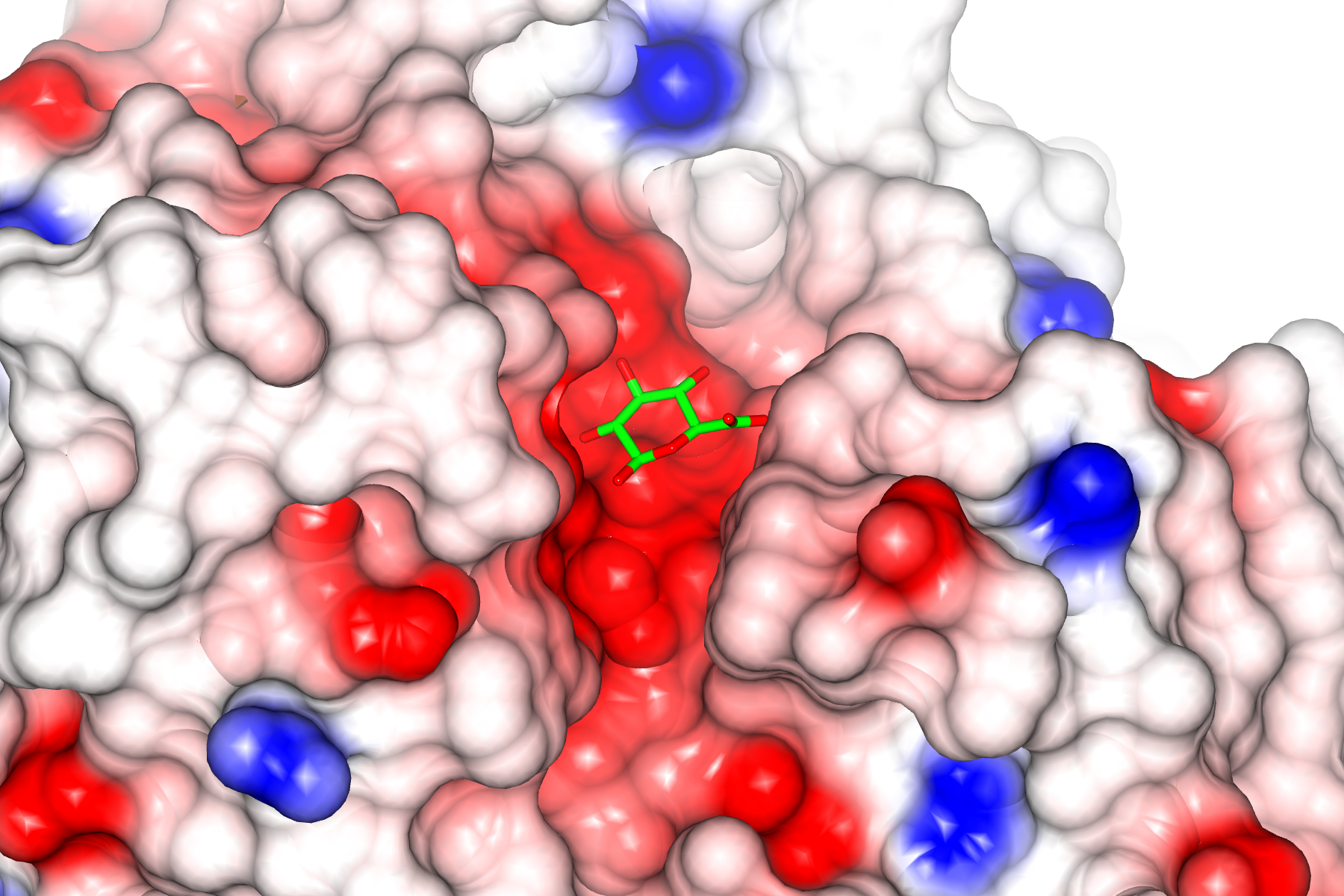
In 2004 Associate Professor Helen Blanchard and her team from the Institute for Glycomics, used X-ray crystallography to determine the structure of the rotavirus carbohydrate recognising domain (VP8*). This protein has the role of recognising host-cell carbohydrates as the initial stage of rotavirus infection. By understanding the structure of the rotavirus deepens the understanding of how the virus attaches to the cell, thereby giving clues as to drugs that could block this process and thereby protect against infection.
This photo essay celebrates the process of X-ray crystallography, from establishing the crystalline structure of the protein through to the X-ray diffraction process and finally mathematically mapping the 3D structure of the protein. All of the images and 3D structures are from Associate Professor Blanchard and her team at the Institute for Glycomics.
To understand how a protein performs biological function, it is essential to know its atomic three-dimensional structure. X-ray crystallography is essentially a form of very high-resolution microscopy. Traditional light microscopes are limited because eventually the wavelength of the light itself is bigger than the object you are trying to see. In this process however, X-rays are used as they have an extremely short wavelength enabling them to be used even down to individual atoms. As the X-rays hit and interact with the electron cloud associated with atoms of the protein they are scattered giving a resulting diffraction pattern. To solve the structure of the protein the pattern of diffracted or scattered X-ray waves is mathematically retraced to reveal location of the atoms that scattered the X-rays.
The Crystals
Only when the protein molecules are packed together in a regular manner, giving a three-dimensional array and forming crystals is one able to use X-ray crystallography to elucidate the atomic structure of the protein. An X-ray beam is passed through the protein crystals and because of the regular packing of the protein molecules the X-rays are scattered in a particular way that generates a diffraction pattern. It is essentially that good quality crystals are generated. Achieving a crystalline structure of the protein can take years of research alone.
Piecing together the structure
Once X-rays interact with atoms within the molecules that are packed in a regular manner within the crystal, they are diffracted. This diffraction pattern can then be mathematically resolved to map the atomic structure of the material. Firstly, clouds are mapped, until ultimately the whole atomic structure of the molecule can be assigned and thus the structure be determined.